PCR (Polymerase Chain Reaction) - a comprehensive overview with tips to help it work well for you!

ฝัง
- เผยแพร่เมื่อ 8 ก.ย. 2024
- PCR (Polymerase Chain Reaction) is a way to amplify (make lots of copies of) specific parts of DNA from larger parts of DNA. You specify the region you want copied using PRIMERS which are short fragments of DNA that bind to where you want the copier (DNA Polymerase) to start & stop. It’s kinda like the original, bigger, DNA template is like a transcontinental railroad & you only want to copy the stretch from Utah to Colorado - the primers act as “train stations” that the DNA Pol “train” travels between, laying down “tracks” (DNA nucleotides) ahead of it as it goes based on the sequence of the other strand.
blog form: bit.ly/pcrtrain
The instructions for making proteins (long chains of amino acid letters that fold up into cool shapes and act as molecular workers) are written in a different biochemical language - DNA. Stretches of DNA that hold “protein recipes” are called “genes” and, since the genetic code is universal, you can stick that gene to “any” cell and that cell’s ribosomes (protein-making machinery) will “understand” it and make the corresponding protein. Similarly, if you know what genes an organism (Including a person) has, you can know what proteins they make, whether they have mutated versions of some genes, how they relate to other organisms.
So, DNA’s really important and kinda like how you might love your IKEA table but don’t have a particular fondness for the instruction manual, but you need it, in order to better understand proteins I often have to deal with their DNA instructions, and to do this I need to make lots of copies of specific stretches of DNA, and thankfully PCR provides a way! The discovery of PCR is attributed to Karry Mullis (working for a biotech firm called the Cetus Corporation) who got the first glimmers of it in 1983 and published it as a legit technique in 1985 (and in 1993 won the Nobel Prize for it).
As with basically all “breakthrough” scientific discoveries, he couldn’t have done it with out the work of countless other scientists working before him. (and we couldn’t do the work we do today without the work of him and others!).
For example, the stage was set in part by Arthur Kornberg, who won the Nobel Prize for discovering an enzyme (reaction speeder-upper) that could copy DNA, using a single strand of DNA letters (deoxynucleotides (dNTs)) as a template for linking up (polymerizing) “opposite” letters into a complementary chain (which can then be used as a template for recreating that original template sequence thanks to the 1:1 “oppositeness” of DNA letters (more below). He called this enzyme DNA Polymerase (DNA Pol) and he could get it to make single complementary copies of DNA, but Mullis’ contribution was using that DNA Pol to make lots and lots of copies.
And we still do. But we typically use super-hardy versions of DNA Pol that can withstand the high temps we cycle them through in the process I’m gonna tell you about. And that choice of polymerase is just one decision you have to (I mean get to) make when you go to the DNA copy machine! So hopefully today’s post will help you iron out your routine. I’m going to start with an overview of how it works (so hopefully not too jargony/wonky) but then I’ll get into some details for those who want them. (apologies in advance for formatting - full-time grad student doing full-time lab stuff…) So let’s go!
DNA (DeoxyriboNucleic Acid) is the biochemical language our genetic info’s written in & its alphabet consists of 4 deoxynucleotide (dNT) “letters,” A, T, C, & G which have a “generic” part made up of a deoxyribose sugar with phosphate(s) hooked up on the “left arm” (5’ position) has a hydroxyl (-OH) group as a “left leg” (3’ position) & then the different letters have different “nitrogenous bases” (bases) that stick off as a “right arm” - these bases are the single- or double-ringed parts.
dNTs use their generic parts to link together through phosphodiester bonds to form long single stranded DNA (ssDNA) & 2 complementary single strands “zip together” using their unique base parts (A across from T, C across from G) to form double-stranded DNA (dsDNA). This double-strandedness protects it from damage (the bases are facing in) and allows for easy copying since if you unzip it one strand can be used as a template for making another.
This “unzip and copy” is what happens in replication - before a cell divides, it needs to copy all its DNA (its entire genome) so that it can pass on a full set to each daughter cell, and it does this with the help of DNA Pol, which brings together the freely-roaming nucleotides, holding the right ones together & helping them link up, while rejecting the wrong ones (ones that don’t complement each other (e.g. don’t let an A bind a C!)
finished in comments


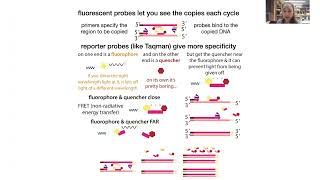






PCR (Polymerase Chain Reaction) is a way to carry out this process in a (really tiny) test tube & only copy a small section of DNA, which we specify by using PRIMERS. Primers are short pieces of DNA (oligonucleotides, or “oligos”) which we design to “bookend” our region of interest (AMPLICON). So, for each reaction we design and order different primers (thankfully they’re cheap!)
We need these primers because DNA Pol can’t start chain-building from scratch - it needs to start from a short double-stranded stretch. This is just one of its limitations (but sometimes limitations can be good! You don’t want cells copying DNA randomly!) Another limitation of its is that it can only copy DNA in One Direction (5’ to 3’). Before we get too far, let’s make sure we’re NSYNC… (90s girl, sorry!)
The letter-linking is “generic” because it only involves the “backbone” parts that all the letters have (phosphodiester bonds involve the phosphate & hydroxyl merging) so you can link letters in any order (e.g. ATTACA or CAAATT). But the strand-zipping is specific because it occurs through interactions of the unique bases. So the “opposite” of ATTACA is TAATGA, which is different from GTTTAA. But, writing the opposites like this is a bit misleading because opposite direction you should be reading.
If you have ATTACA, and you stick the complementary letters across from it, you get this:
ATTACA
TAATGA
BUT - dsDNA is ANTIPARALLEL - this means that the strands are running in opposite directions (one is 5’->3’ and the other is 3’->5’ with the ‘ pronounced “prime” and referring to whether the left arm (5’) or “left leg” (3’) of that end’s sugar is free). So
5’ ATTACA 3’
3’ TAATGA 5’
And we usually write sequences 5’ to 3’, so the “complimentary sequence” to 5’ ATTACA 3’ is 5’ AGTAAT 3’
This may seem like a mere technicality, but it’s really important in reality! Because DNA Pol can only copy DNA in one direction, 5’ to 3’, and you always have to keep in mind which way the “train tracks run”
Train tracks? This is another weird analogy of mine - I like to think of nucleotides as train tracks and DNA Pol as a train. This train can only travel on double-stranded track, so it has to lay the track down ahead of it as it goes (and it knows what track to lay down by making it “match” the other side of the track (e.g. if the next track across from it (on the other strand) is a T, lay down an A). In PCR, primers provide the starting stations for the train (since DNA Pol needs double-strandedness to start, it’ll only start where you make it double stranded (but shorter than the other strand so there’s still stuff to copy. Basically, you want something like this:
--====-- to make this --========
PCR is run in cycles of 1⃣ MELT (heat up dsDNA to unzip strands) 2⃣ ANNEAL (cool down slightly to allow primers to bind and 3⃣ EXTEND (starting where primers leave off, add nucleotides complementary to template strand until you reach end of template strand). After the 1st cycle (where Pol goes till it runs out of steam or out of time), this end is determined by other strand’s primer because DNA can only *copy* it cannot “compose” so it’ll run off the track corresponding to the position that strand started being copied from in the 1st round. (easier to explain in pics)
You do this over & over 🔁 (30 or so times) to get lots of copies (each time you get 2X as many copies because each new strand becomes another template strand).
It might sound like a ton of work - and for the molecules it is! - but for us, once we get the reaction set up, the hardest part is just waiting for it to finish! Not for Mullis, however… It’s “easy” these days because we have machines called thermal cyclers that do the rapid heating and cooling and heating and cooling and heating and cooling and…
But these machines didn’t come on the scene until 1987 (and of course not every lab could afford them, etc.) so in the beginning scientists would manually transfer the reaction tubes from one water bath to another over and over again.
The reaction to link nucleotides together (nucleotide polymerization) is the same regardless of whether it’s happening in your cells or in the tube. It’s also the same for RNA (but using a different Pol). but here we’ll speak in DNA terms. So the reaction takes 2 deoxynucleotide triphosphates (dNTPs)(have 3 phosphate (PO₄³⁻) groups linked together) and links them up. And when it does so, it kicks 2 of those phosphates as a molecule pyrophosphate (PPi)
So, same process “in vivo” (in the body) and “in vitro” (in an artificial setting like a test tube), BUT in cells there are lots of helpers, whereas in PCR the process is stripped down to its bare necessities:
🔹TEMPLATE DNA: dsDNA containing the sequence you want to copy (amplify)
🔹PRIMERS: short ssDNA complementing the ends of amplicon (1 to serve as a start site for each strand)
🔹dNTPS: nucleotide building blocks (letters) to be added
🔹BUFFER: liquid combo of salts & pH stabilizers to keep everything happy
🔹Mg²⁺: magnesium cation (➕ charged molecule) to act as “chaperones” to help shield phosphates’ ➖ charge
🔹and, drumroll please… 🥁🥁🥁 DNA POLYMERASE (DNA Pol)
One of the hardest parts of carrying out a biochemical reaction is often bringing reactants together & keeping them together long enough to interact. Why’s this so hard? Molecules like to be free to move around (they want high ENTROPY) & they don’t like to be tied down. Entropy refers to how many different “states” a molecule can be in (this can refer to being in different places or in slightly different shapes (e.g. bond rotated a bit). It’s sometimes described as “randomness” or “disorder” because the more ways something can move the less likely you are to know exactly where they are at any time.
Getting nucleotides to link up is like getting a bunch of kids running around at recess to link up to form a really long 3-legged race. First, you have to get them to come over, then you have to get them to stay still long enough to convince them to link up, and then once they’re linked up you have to make it “fun” for them even though they can’t run around anymore because their movement is restricted by being linked to the person next to them. And Pol has an even harder time because it has to get the kids to link up in a specified order!
So how can one little protein do all this? By giving the nucleotides something they want in return - ridding them of a couple of their phosphates. The phosphates stick off the 5’ end & they’re basically really concentrated negative charges clamped together like a spring. PHOSPHATE (PO₄³⁻) has a central phosphorous(P) atom connected to 4 oxygen(O) atoms. It has “extra” electrons (e⁻) so it’s ➖ charged. Like charges repel, so phosphates don’t like to be next to each other. So it takes effort (in the form of energy (E)) to bring & hold phosphates together (like compressing a spring) - so we call these bonds “high energy” and when they’re broken apart that E’s freed to be used for other things like paying cost of linking nucleotides together.
So, even though nucleotide polymerization is still energetically costly because you’re tying down molecules, ⬇ their freedom (⬇ entropy), this is compensated for by the large ⬆ in entropy that occurs when PPi is released & hydrolyzed (split by water) to give you 2 individual orthophosphates (Pi) (and 2 little things moving around freely has even more freedom than the 1 medium-little thing (PPi) that’s initially released - so you get a double-boost
BUT in order to get this benefit, you 1st need to get them to react & this is often where proteins &/or RNA CATALYSTS (reaction speeder-uppers) called ENZYMES come in. They act as a sort of “mediator” bringing the right reactants together, holding them in optimal positions to react, stabilizing reaction intermediates. & providing a friendly environment.
As I mentioned before, Arthur Kornberg discovered some enzymes that help mediate DNA copying: DNA POLYMERASES (DNA Pols). DNA Pol helps hold an incoming nucleotide (of the triphosphate variety) close to the growing chain it needs to be added to & in the right position. The 3’ hydroxyl (OH) group then goes in for the attack! It latches on to the 1st phosphorus (P) group of the incoming nucleotide -> that P now has too many bonds, so it kicks out the other 2 phosphate groups as the inorganic phosphate molecule PYROPHOSPHATE (PPi) (energy boost 1). PYROPHOSPHATE is then hydrolyzed (broken by the addition of water) into 2 molecules of ORTHOPHOSPHATE (Pi) (energy boost 2).
DNA Pols have that general mechanism in common, but different organisms have slightly different DNA Pols that vary in EFFICIENCY, PROCESSIVITY, FIDELITY, & THERMOSENSITIVITY
🔹EFFICIENCY: how fast can it go?
🔹PROCESSIVITY: how many nucleotides can it add before it falls off template?
🔹FIDELITY: how many typos does it make?
🔹THERMOSENSITIVITY: how much heat can it take?
You can get higher FIDELITY (fewer typos) if your Pol has a “proofreading” 3′→ 5′ exonuclease (DNA end-chewing) domain that can sense errors, “backspace” to remove them, & then put in the correct letter. This profreedng is important because errors will get copied… & copied… & copied… BUT it slows down process so you get lower EFFICIENCY.
Other parts of DNA Pol proteins can also help out. A way to increase EFFICIENCY is by increasing PROCESSIVITY - keep Pol on the template. Constantly falling off & hopping back on surely slows you down! Processivity-enhancing domains (a “domain” is just a protein “section”) or separate processivity-enhancing “subunits” bind dsDNA to help latch Pol on. But importantly they don’t bind “too tightly” & they can bind any sequence - this allows Pol to stay on but slide along
BUT before you can copy strands you have to unzip them & this too is energetically expensive (like peeling apart 2 pieces of stuck together tape). You have to put in energy to give the DNA molecules more energy so they wiggle around more & the strands come apart.
In your cells, enzyme helpers called HELICASES help unzip them using chemical energy from ATP, but our “bare-bones” PCR version doesn’t have these helpers. Instead, we get the needed energy from HEAT. In the MELT step, we physically heat up the dsDNA so the strands come apart. And we have to get it REALLY hot! (~95°C or 200°F). Human DNA Pol would be pretty useless at this temp bc same heat that causes strands of DNA to come apart (yay!) can also cause proteins to unfold (eek!) (just like chains remain chains when you melt DNA (you don’t break up strong covalent bonds), heat denaturation of proteins leaves you w/chains of amino acids)
Thankfully there are organisms called THERMOPHILES that have evolved to live in super hot environments (like near thermal vents in the ocean). They have super-strong proteins that can withstand high temps needed PCR. The “classic” PCR Pol is Taq, which was discovered in 1976 and gets its name because it comes from the thermophilic bacterium Thermus aquaticus. Taq really made PCR possible - before that, scientists trying to copy DNA in the lab were using a DNA Pol from e. coli bacteria. That DNA Pol couldn’t take the heat, so after each heat step, they’d have to add more! In fact, the first thermal cycler, named “Mr. Cycle” was designed with an open system so you could keep adding more. bit.ly/2FoUFkD
So Taq was a major, crucial, discovery. But it’s definitely not “perfect” - Taq tends to make a lot of typos (low FIDELITY). Pfu DNA polymerase (from Pyrococcus furiosus) makes fewer errors (so higher FIDELITY). BUT it has relatively low EFFICIENCY. Could we do better?
It’s hard to get all 4, but that hasn’t stopped scientists from trying! Scientists can stitch together parts they like from different Pols to get Pol “chimeras” w/enhanced functions. Our lab uses a chimera called Phusion Polymerase (not a paid endorsement, just what we use!) It’s based off of a Pfu-like DNA Pol (w/proofreading capability for increased fidelity) fused to a small dsDNA-binding protein called Sso7 (from Sulfolobus sulfactaricus) which serves as a processivity-enhancing domain. It can add 1000 nucleotides (1 kb) in only 15 seconds w/few errors! So we time out the extension step accordingly (e.g. if we want to copy a 4kb segment, we’ll set the extension step for 4x15=60s
So, DNA Pol was one choice we need to make, but there are others too….
Where what do you want your start & stop sites to be? Design primers to match - but not just any sequence… It’s really important to design the stations (primers) carefully. Like many things in biochemistry, it’s largely a matter of AFFINITY & SPECIFICITY
SPECIFICITY. We want the primers to bind ONLY where we want them to bind. Say you ask some friends to buy you a train ticket for a trip from Kansas City to NYC Is that Kansas City, Kansas? Or Kansas City, Missouri? Some friends might think Kansas, others might think Missouri & you end up with 2 types of train tickets..
Similarly, if your primer can bind at multiple sites on the DNA, you end up copying different stretches of train track giving you a mix of “nonspecific products” which show up as multiple bands on an agarose gel you use to separate the DNA pieces by size & visualize them: bit.ly/agarosegelrunning
When you design primers, you want to make sure they match your site of interest & ONLY that site. Like a computer password, the longer the sequence, the more likely it is to be “unique.” If there are multiple occurrences of sequence you initially choose you might have to lengthen it to include more of the surrounding sequence (like saying “find a blue house w/a red house on the left & a green house on the right” instead of just saying “find a blue house”) You can use free software programs like NCBI BLAST or Primer3 to help you check for specificity & design good primers
BUT too long a primer and you can face other problems that lead to a “PCR TLDR” (too long didn’t read)…
🔹 PRIMER DIMERS: this is where the primer binds to itself instead of to your template. These can be self-dimers (where 2 “start stations” or “stop stations” bind to themselves or cross dimers (a start & a stop)
🔹SECONDARY STRUCTURE: a single primer can fold up into “hairpins” & bind itself
This leads to less primer available to bind template, so lower yield (less copies made) & DNA Pol can end up using primers as a (really short) template, amplifying primer “artifacts” instead of desired amplicon. And the high primer concentrations needed to prevent template-template zipping, make such primer pairing more likely because there are more primer fish & fewer template fish in the sea
Secondary structure in the *template* can also be a problem - some regions of DNA are tightly wound up, making it hard to get to the site to bind (it’s hard to build a train station in the middle of a mountain pass). You also want to avoid repetitive stretches (things like “AAAAAA”) because it makes it easier to “slip” & misprime
Typical primers are usually ~20 nucleotides (nt) long, but it depends on experiment type, etc. There’s free software available (like NCBI Primer-BLAST, Primer3, & AmplifiX) to help you design primers to fit your needs.
And remember - get the reverse compliment! bit.ly/sequencetermstools
⠀
AFFINITY: We want the primers to have high affinity (attractiveness & stickiness) for the site we want them to bind so that they’ll bind there stably & not fall off randomly during the annealing or extension steps. BUT we don’t want the affinity to be too high or it won’t come off during the melt steps
Affinity is largely dependent on the primer length (longer primers have more interstrand bonds working together to keep the strands glued shut) & “base composition,” When G’s & C’s are across from each other they can form 3 H-bonds. But when A’s & T’s are across from each other they can only form 2 H-bonds, so G-C pairs are stronger than A-T pairs (and G-C rich regions have stronger base-stacking interactions). So a higher “G-C” content (typically given as a % of bases) means stronger binding. Ideal is usually ~40-60%
note: this is why origins of replication (ORIs) (where DNA strands come apart to be copied before cells divide) tend to be “A-T rich” because it makes it easier to melt them apart
Just like it’s easier to pull off a piece of tape from the end than the middle, it’s easier to pull of primers from the end, so you might want to put a“G-C” clamp at the 3’ end (1 C or G) to help latch it on tight
A common measure used to calculate/report this affinity is the Tm (melting temperature). It’s the temperature at which 1/2 the primer is bound - the higher the temp, the more energy the molecules have & the harder it is to get the DNA to “stay still” & bind. A high Tm means the affinity’s high enough to hold down the wriggling DNA. Sp higher Tm, higher affinity. You typically want ~60°C & you want the Tm of the 2 primers to be similar to one another (within ~5°C)
You want to know these Tms because they’ll help you decide what temp to use for your annealing temperature - they temperature you program the thermal cycler to be at during the primer-binding step of each cycle.
How do decide? The higher the temp, the more energy the DNA molecules have so it’s harder for them to be “tied down.” At higher temps, they have to really like their binding partner in order to sacrifice the freedom to move freely. But at lower temps, they have less energy, so they’re less “picky” & more likely to “settle” for less-optimal pairing. As a result, if the annealing temp is too low, your primers can “misprime” & bind at the wrong sites giving you a mixture of “nonspecific” products.
So you want to choose a Goldilocks ANNEALING TEMPERATURE where the DNA molecules have enough energy to seek out their soulmate, BUT not so much that they can’t “tie the knot” once they find it. To help you choose, you can use free software to calculate the melting temperature (Tm) of the primers based on their length & sequence (since G’s & C’s bind each other more strongly than As & Ts (thanks to their 3rd H-bond), higher GC content increases the Tm. Common annealing temps are ~55-80°C, and you want the Tms for both primers to be similar so that they both get to be Goldilocks-happy at the same time.
How long should the extension step be? Basically, each cycle, you need to give DNA Pol enough time to copy the region between the primers. So the optimal time depends on length of the sequence you want copied (AMPLICON SIZE) & the speed (efficiency) of the DNA Pol. The “classic” Taq polymerase (a DNA Pol that can tolerate the high temps required) can add ~360nt/min, but “newer models” (either from other organisms &/or mutated) like Pfu Turbo can go faster (~1000nt (1kb)/min)) So, for example, if you’re using Pfu Turbo to copy something 2kb long, you’d want to make sure your extension is a little over 2 minutes. This is per cycle so with longer times, be prepared to do some waiting (a great time to pour that agarose gel you’ll use to check that it worked!)
Just how long you’ll have to wait depends on how many cycles you choose. And that depends in part on how many copies you want made (each cycle you increase # of copies exponentially (e.g. 2, 4, 8, 16, 32, 64…), so by 35th cycle you’d have 68 billion copies! You might think, the more the better, right? BUT the more copies you make, the more chance for errors to occur (& be copied) Common cycle #s are ~25-30 (but even if you have to wait you don’t have to manually move the tube over and over!)
more on DNA sequencing: bit.ly/DNAsequencingmethods
more about all sorts of things: #365DaysOfScience All (with topics listed) 👉 bit.ly/2OllAB0 or search blog: thebumblingbiochemist.com
I just started volunteering in a microbiome lab. This video is helpful. Do you share your slides? Thanks!
Thanks! I would but I'd have to find these specific ones and upload them and am swamped with work currently. Sorry!
@@thebumblingbiochemist no worries. keep fighting the good fight. thank you for your content