hello, i find molecular dynamics hard to learn as i had no prior experience with coding and linux (what my teacher recommended me to use), videos like this makes it very easy for me to understand the concept and how to do it. THANK YOU FOR THE VIDEO!
Sir, at 9:25 you add Kollman charges to the protein. However, at 15:23, when you load the protein macromolecule, you click on 'no' for preserving Kollman charges. The next popup box states that Gasteiger charges were added. So, should we add Gasteiger charges for the protein?
I'm in bsc 2nd year and I could understand anything sir ... I'm just curious why did you remove the water molecules and add polar hydrogen to the protein ??
I'm getting this error. Can anybody help me clear this out? Thank you. Traceback (most recent call last): File "C:\Program Files (x86)\MGLTools-1.5.7\lib\site-packages\ViewerFramework\VF.py", line 941, in tryto result = command( *args, **kw ) File "C:\Program Files (x86)\MGLTools-1.5.7\lib\site-packages\AutoDockTools\autoflexCommands.py", line 259, in doit dict = self.vf.flexDict AttributeError: MoleculeViewer instance has no attribute 'flexDict'
Im performing docking on a known active site so i limited the gridbox around the active site only. Some of the conformations i got after docking were outside the grid box. Is that normal?
Hi thanks for this, came at the right time! When adding the grid box, is it generated automatically at the binding pocket or will have to adjust it? if so, how do i ensure its the 'correct' coordinates?
Is it possible to do docking without keeping the the AD4.1_bound.exe, AD4.1_parameters.exe and autodock4.exe, autogrid4.exe files in the same working folder?
@@saikatmandal929 I understood that you don't have to use vina.exe but directly autogrid4 and autodock 4 on autpdock tool. But when I try to put the new parameters the program doesn't run
Thank you sir for such a nice video. One question- if we don't know the active site, why we are not enclosing the whole protein inside the grid by changing dimensions.?
Could you make a video showing how to dock in Vina Auto-docking using some flexible active site waste? I need my molecule to interact with specific residues and I don't know how to do it. Please.
Hi joyce I think you cover all the protein by doin grid box so it will be flexible as you took all protein whereever will get less energy it will enter
As we all know that docking simply interaction between receptor and ligand molecule, before docking we do remove the water molecule and some other additional ligands molecule , and add polar hydrogen bond also, Afer that we dock between receptor and ligand molecules, and more negative sign give the good stability.....but sir my question is that in natural condition how water and additional ligands remove from receptor molecules and how will i add polar hydrogen also?
i tried to download autodock vina using the above mentioned link but it is appearing as page not found.... please help. can please anybody help me in downloading it
Hi, I have a issue. I'm not able to get docking results as it show error like, FATAL ERROR: ERROR: 3778 records read in, but only dimensioned for 2048. Change "MAX_RECORDS" in "constants.h". Can u please suggest how to rectify this error ?
Sir can we docked two target site for same disease by single ligand? Sir suppose x naam kaa ek ligand h jo A naam ke target site ko inhibit krra ho , phir suppose wahi x naam kaa ligand B naam ke target site ko inhibit krra ho, to sir kya hum ek saath A ligand ko dono target site(A and B) pe ek he saath docking kra skte h..... Multiple target site docking.... Sir plz doubt clear kar dijiye Agar kar sakte hain to kese ? Nahi kar sakte hain to kyu ni kr sakte?
Autodock tool can dock one protein-ligand at a time, its how any docking tool is designed. So you have to perform docking of 2 ligands with a protein then you have to perform docking twice.
At 17:55, Spacing parameter is set 0.375 A, but this is not used by AutoDock VINA. Changing this parameter affects the grid box size. So even you attempt to perform site specific docking, the docking is going to be blind docking due to this parameter. I am not able to get the clarification about this parameter while docking except VINA official tutorial has this option set as 1.
Hi Dr. Sanket. I am an IBDP 2 student. For my school project I need to conduct multiple ligand simultaneous docking. I was able to conduct single ligand docking but am unable to understand how to do MLSD. Can you please help with instructions on how to do this. Thank you
Sir suupose hum ek research article likh rahe hain , sir kya hum comparison ke liye aesa kar sakte hain kya, jese remdesivir ke liye dimension xyz alag rakh rahe hain isme humne docking autodock vina , command prompt waale step se docking ki , or other multiple ligands ke liye humne autodock vina , command prompt se docking nahi ki more than two ligands hone ke usse , isme humne pyrx virtual screening ka use kiya or dimension xyz bhi different rakhi as compare to remdesivir... To sir kya hum comparison kar sakte hain ab remdesivir or ye multiple ligands kaa.... Yaa hume sabhi ligands ke liye pyrx virtual screening he krni pdegi , agar hum research article m isko publish karana chahte hain to.... Sir koi problem create to nahi hogi dimension different rakhkr comparison kiya karkr
Hello sir, I'm from bioanalytical Sciences ... We had your workshop that I couldn't attend because of personal problems n we have this docking by autodock vina in our practicals n we have our practicals from 8 ... N I'm having some major issues... Whenever I try to download protein it gets downloaded in rasmol raswin than pdb extension .... N ligand in sdf where it supposed to be downloaded in MDL sdf format n because of it I'm not able to proceed ... Can you please suggest me what can be done ?
I have a doubt. I guess The grid box has to be formed around the active site residues by changing the x, y and z axis values so that the interaction can be calculated correctly,
Sir, I downloaded autodock-vina from the link but after installing the program, it does not have the brown ribbon you used to save the protein and ligand, what should I do?
i use pyrx for docking, and use multiple ligands i download them from zinc database (nubben database for natural compounds), i do minimization and docking but when finish the result are encodede for the compound it just codded the energy , and they are 2332 compound how can i know the compound crosspend to each result???
hii.. I need your help. everytime I am trying to save as pdbqt format, my file is being saved as pdf.. could you please tell how to resolve this problem
my protein doesnot contain cl atom which is there in the ligand and maps are generated for all atoms in the protein. while autodocking its showing the error: I'm sorry; I can't find or open "5nm2.Cl.map" the atoms in protein are : A C H HD N OA SA and atoms in ligand are:A Cl OA N pls tell how remove the error
sir same dimensions rakhne main result alag alag kyu aata hai, jese maine ek ligand ke liye dimension x, y, z 30,40,50 rakhi or binding energy 10 aare h autodock vina se, but jab main yahi dimension rakhta hu same lifand ke liye, par is time main pyrx virtual screening tool se karta hu to result 9 binding energy kyu aata hai
can anyone please explain my following question? this particular protein "4mzi" in this video has some missing residues in its side chain. so is it possible to do docking without fixing it ? in this mgl tool he didnt fix the missing residue. will that make good docking ?
Hello sir, when i click on grid and selecting the protein then get error, showing protein pdbqt has one or more hydrogens with no bonds. Plz solve this problem
Hello Sir! thank you for your videos,i have a question ,when i drug the pdb files into Autodocktools software,it occurs a error “4mzi.pdb does not exists”,hope you can help me to solve this problem,thank you!
why you have not deleted zinc metal in the protein during protein preparation,any reason behind this??? If i delete any problem ? can you explain? I shall be thankful to you
Sir.. Everything is useful in this video. But am unable to download PyMOL. My system can't allow that to instal. So, kindly send the ligand format file, sir.
Sir few docking files in the folder remain hidden and no matter what I do, I'm not able to view them in the Docking folder. It's visible in AutoDock when I select Read Molecule but not in the folder. How do I resolve this sir?
Respected sir need help The command shows the program is not recognized as an internal or external command operable program or batch file what is the solution for this problem
Hi! very helpful video indeed. I have one query which is my SDF file is not getting recognized as a SQLCE database. how do i overcome this error? Kindly help..
Hi Sanket in Ur video you have not mentioned the path of folders where we must keep the files to run autodock, when I am running auto dock it showing error
If I use a receptor that I have already known the binding site with a reference ligand, how can I set the grid box in that binding site only for docking with other molecules?
Thank you Sanket Bapat. Both the videos are very neatly and clearly presented. Highly informative. But once after docking how are we to interpret it in the paper. Any suggestions or advice regarding the same.
Good day to you sir. Thank you for this excellent interactive session. I have a query, What is to be done if ligand profiles are not seen in the downloaded material from PDB.



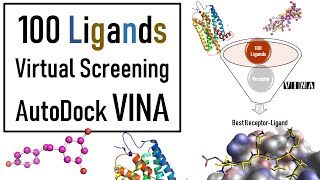






thank u Sanket B.. excellent and more useful both of docking videos
Thank you very much!
I think I'll put your name in acknowledgment, you are the only one who helped me in learning which I was roaming around behind everyone... Thanks
Hello Sir! How to prepare script for multiple ligand docking?
Yes please!
i want to give you the credit if I got a paper published like literally ...Extremely helpful
hello, i find molecular dynamics hard to learn as i had no prior experience with coding and linux (what my teacher recommended me to use), videos like this makes it very easy for me to understand the concept and how to do it. THANK YOU FOR THE VIDEO!
Uyhv4eèhj
Đ
ŘGGĎDGGTR7
Why do you skip some steps like energy minimization of ligand and protein and charges added to ligand
Can anyone help me out with the installation , I seem to have problem with finding the vina folder in the path hence not able to attempt docking
Sir, at 9:25 you add Kollman charges to the protein. However, at 15:23, when you load the protein macromolecule, you click on 'no' for preserving Kollman charges. The next popup box states that Gasteiger charges were added. So, should we add Gasteiger charges for the protein?
It says could not open "config.txt" for reading while clicking enter from (8:35). Please help
I'm in bsc 2nd year and I could understand anything sir ... I'm just curious why did you remove the water molecules and add polar hydrogen to the protein ??
I'm getting this error. Can anybody help me clear this out? Thank you.
Traceback (most recent call last):
File "C:\Program Files (x86)\MGLTools-1.5.7\lib\site-packages\ViewerFramework\VF.py", line 941, in tryto
result = command( *args, **kw )
File "C:\Program Files (x86)\MGLTools-1.5.7\lib\site-packages\AutoDockTools\autoflexCommands.py", line 259, in doit
dict = self.vf.flexDict
AttributeError: MoleculeViewer instance has no attribute 'flexDict'
These two videos have been really helpful for my work. Thank you so much.
like your tutorial video sir, very helpful.
Thank you so much 😇🙏
you are doing grat job. Could you prepare a molecular docking video for ligand-based drug design.
Im performing docking on a known active site so i limited the gridbox around the active site only. Some of the conformations i got after docking were outside the grid box. Is that normal?
i am looking for PDB file of Beta Amyloid Protein (Alzheimer's Disease) can anybody help please.
I have done docking in vina by coding only..autodock is GUI, isn't Vina acessesd by coding only?
Very useful both parts for autodock vina learning
Can you make a video about how to minimize energy please
सर मला तुमचा contact no द्या, मी मुंबईत phd करतोय, मला हवंय हे docking
Can anyone plz help me for my project am now in final year of B pharm ..I want to take project on docking
sir I wanted to know..if the same thing can be executed in ubuntu system
My pdb file is getting downloaded in the form to words how can I get in form of image
I love this. It is so helpful and explanatory.
my protein file is not saveing as the pdbqt so what should i do
I am not getting the grid option in my Python molecule viewer. Can anyone tell me, how to get it?
Sir, Amber 18 pe kaise simulation hota hai....iska video banaiye kabhi 🙏
Hi thanks for this, came at the right time! When adding the grid box, is it generated automatically at the binding pocket or will have to adjust it? if so, how do i ensure its the 'correct' coordinates?
from literature you have to search
Is it possible to do docking without keeping the the AD4.1_bound.exe, AD4.1_parameters.exe and autodock4.exe, autogrid4.exe files in the same working folder?
Yes its possible, even my working folder doesn't have the executable file
You are doing a very good job. Keep it up Dr.
Auto dock vina or auto dock 4 different h.. Lakin unka page same kiun h..koi bta skta h..
Sir, very nice video
Can you please make a video in which you explain how adding a metal ion ( in my case is tin)
I am facing problem for adding pt metal.
@@saikatmandal929 I understood that you don't have to use vina.exe but directly autogrid4 and autodock 4 on autpdock tool. But when I try to put the new parameters the program doesn't run
Thank you sir for such a nice video. One question- if we don't know the active site, why we are not enclosing the whole protein inside the grid by changing dimensions.?
Good question, yes we can encompass the whole protein.
Could you make a video showing how to dock in Vina Auto-docking using some flexible active site waste? I need my molecule to interact with specific residues and I don't know how to do it. Please.
Hi joyce I think you cover all the protein by doin grid box so it will be flexible as you took all protein whereever will get less energy it will enter
great video content about docking
Can DNA-Protein docking be performed in Autodock? Please suggest a suitable tool for building DNA 3D structure
Amazing it's educating thanks
As we all know that docking simply interaction between receptor and ligand molecule, before docking we do remove the water molecule and some other additional ligands molecule , and add polar hydrogen bond also, Afer that we dock between receptor and ligand molecules, and more negative sign give the good stability.....but sir my question is that in natural condition how water and additional ligands remove from receptor molecules and how will i add polar hydrogen also?
i tried to download autodock vina using the above mentioned link but it is appearing as page not found.... please help. can please anybody help me in downloading it
Made changes, Thank you!
I never thought i'll enjoy learning simulations so much. Thank you for making it so simple and easy. :)
What about chimera or autodock and pymol is more effective...thank so much Dr.sankat🎉🎉😊
Hi, I have a issue. I'm not able to get docking results as it show error like, FATAL ERROR: ERROR: 3778 records read in, but only dimensioned for 2048.
Change "MAX_RECORDS" in "constants.h".
Can u please suggest how to rectify this error ?
Sir can we docked two target site for same disease by single ligand?
Sir suppose x naam kaa ek ligand h jo A naam ke target site ko inhibit krra ho , phir suppose wahi x naam kaa ligand B naam ke target site ko inhibit krra ho, to sir kya hum ek saath A ligand ko dono target site(A and B) pe ek he saath docking kra skte h..... Multiple target site docking....
Sir plz doubt clear kar dijiye
Agar kar sakte hain to kese ?
Nahi kar sakte hain to kyu ni kr sakte?
Autodock tool can dock one protein-ligand at a time, its how any docking tool is designed. So you have to perform docking of 2 ligands with a protein then you have to perform docking twice.
Thanks for the amazing explanation
In docking...grid macromolecule protein are choose..then show error....so, what do i do now....
Plss..help me..
At 17:55, Spacing parameter is set 0.375 A, but this is not used by AutoDock VINA. Changing this parameter affects the grid box size. So even you attempt to perform site specific docking, the docking is going to be blind docking due to this parameter. I am not able to get the clarification about this parameter while docking except VINA official tutorial has this option set as 1.
Hi Dr. Sanket. I am an IBDP 2 student. For my school project I need to conduct multiple ligand simultaneous docking. I was able to conduct single ligand docking but am unable to understand how to do MLSD. Can you please help with instructions on how to do this. Thank you
Can I get the part 2 link please
Is there any rule regarding which chain should be kept and which one should be deleted ?
Sir suupose hum ek research article likh rahe hain , sir kya hum comparison ke liye aesa kar sakte hain kya, jese remdesivir ke liye dimension xyz alag rakh rahe hain isme humne docking autodock vina , command prompt waale step se docking ki , or other multiple ligands ke liye humne autodock vina , command prompt se docking nahi ki more than two ligands hone ke usse , isme humne pyrx virtual screening ka use kiya or dimension xyz bhi different rakhi as compare to remdesivir...
To sir kya hum comparison kar sakte hain ab remdesivir or ye multiple ligands kaa.... Yaa hume sabhi ligands ke liye pyrx virtual screening he krni pdegi , agar hum research article m isko publish karana chahte hain to.... Sir koi problem create to nahi hogi dimension different rakhkr comparison kiya karkr
Sir, can we used ligand file prepared in autodock tool in Vina and what about energy minimization step?
Hello sir, I'm from bioanalytical Sciences ... We had your workshop that I couldn't attend because of personal problems n we have this docking by autodock vina in our practicals n we have our practicals from 8 ... N I'm having some major issues... Whenever I try to download protein it gets downloaded in rasmol raswin than pdb extension .... N ligand in sdf where it supposed to be downloaded in MDL sdf format n because of it I'm not able to proceed ... Can you please suggest me what can be done ?
Thank you very much, well explained. Is the Auto dock vina App free?
I have a doubt. I guess The grid box has to be formed around the active site residues by changing the x, y and z axis values so that the interaction can be calculated correctly,
Sir, I downloaded autodock-vina from the link but after installing the program, it does not have the brown ribbon you used to save the protein and ligand, what should I do?
Cyp 17 ko pyrx virtual screen tool main upload karne se alternate conformation bata raha hai, or pdbqt file main convert nahi ho raha hai....
i use pyrx for docking, and use multiple ligands i download them from zinc database (nubben database for natural compounds), i do minimization and docking but when finish the result are encodede for the compound it just codded the energy , and they are 2332 compound how can i know the compound crosspend to each result???
hii.. I need your help. everytime I am trying to save as pdbqt format, my file is being saved as pdf.. could you please tell how to resolve this
problem
my protein doesnot contain cl atom which is there in the ligand and maps are generated for all atoms in the protein. while autodocking its showing the error: I'm sorry; I can't find or open "5nm2.Cl.map"
the atoms in protein are : A C H HD N OA SA and atoms in ligand are:A Cl OA N
pls tell how remove the error
sir same dimensions rakhne main result alag alag kyu aata hai, jese maine ek ligand ke liye dimension x, y, z 30,40,50 rakhi or binding energy 10 aare h autodock vina se, but jab main yahi dimension rakhta hu same lifand ke liye, par is time main pyrx virtual screening tool se karta hu to result 9 binding energy kyu aata hai
PLZ MAKE A VIDEO ON CABS DOCKER
I was done project work the same topic Sir.
can anyone please explain my following question? this particular protein "4mzi" in this video has some missing residues in its side chain. so is it possible to do docking without fixing it ? in this mgl tool he didnt fix the missing residue. will that make good docking ?
I have installed autodock vina, but it is not working. would you please share the alternate link?
Hi! Tab with Ligand icon not available to do ligand preperation in MGL tool. Appreciate your help regarding this how to go about
Thank you Sir
It's very helpful can make a tutorial or multiple ligand docking and results interpretation on auto dock instead of pymol
It´s so nice your video, Sr! Thank you
Sir grid option is not given in my software...What to do now plz tell anyone...??
Hello sir, when i click on grid and selecting the protein then get error, showing protein pdbqt has one or more hydrogens with no bonds. Plz solve this problem
Honestly I appreciate so much your video thank you
Drag and drop is not working for me can you please help
Try File and Open the desired structure
Sir please can u name any inhibitor for hgd gene for alkeptonuria disease
Hello Sir! thank you for your videos,i have a question ,when i drug the pdb files into Autodocktools software,it occurs a error “4mzi.pdb does not exists”,hope you can help me to solve this problem,thank you!
why you have not deleted zinc metal in the protein during protein preparation,any reason behind this??? If i delete any problem ? can you explain? I shall be thankful to you
Sometimes metal ions can play significant role in docking, so when you consider a protein for docking know whether ions are important or no
Sir.. Everything is useful in this video. But am unable to download PyMOL. My system can't allow that to instal. So, kindly send the ligand format file, sir.
Sir few docking files in the folder remain hidden and no matter what I do, I'm not able to view them in the Docking folder. It's visible in AutoDock when I select Read Molecule but not in the folder. How do I resolve this sir?
Very nice information Dr. Gajanan Dongare Akola Maharashtra
It is necessary in autodock to kept the torsion agnles of ligand less than 6?
Thanks for great tutorails! Small question - why do not you add all hydrogens but only polar ones?
Hbb 147***
Can you please let me know why do we add polar hydrogens and kollman charges?
Before preparation both protein and ligand minimize step is important
Respected sir need help
The command shows the program is not recognized as an internal or external command operable program or batch file
what is the solution for this problem
if I follow the step by step of your docking lecture, then can i make the HLA/HLA-DBR and epitope docking properly for vaccine design??
Hi! very helpful video indeed. I have one query which is my SDF file is not getting recognized as a SQLCE database. how do i overcome this error? Kindly help..
Sir thank you
Hi Sanket in Ur video you have not mentioned the path of folders where we must keep the files to run autodock, when I am running auto dock it showing error
If you see the video I had created the folder on Desktop, you can create your destination folder anywhere you want.
😁
If I use a receptor that I have already known the binding site with a reference ligand, how can I set the grid box in that binding site only for docking with other molecules?
you are just pathfinder for bioinformatics learner.
Kindly heed.
Sir,can you tell how to determine ki for inhibitors using autodock
hello Dr Sanket. after running vina, Error: could not open "ligand.pdpqt" for reading. ligand is mol2 file format. how to turn it into pdpqt format?
not being able to save as pdbqt? gets saved as pdb? why?
Good, but need more clarity in explanation
Very well explained .. sir thankyou so much for ur efforts..
Thank you very much!
Thank you Sanket Bapat. Both the videos are very neatly and clearly presented. Highly informative. But once after docking how are we to interpret it in the paper. Any suggestions or advice regarding the same.
Can we perform docking with metal complexes ( Pt complex)?
Good day to you sir. Thank you for this excellent interactive session. I have a query, What is to be done if ligand profiles are not seen in the downloaded material from PDB.